
Síndrome de Waardenburg
- 26 de out. de 2018
- 2 min de leitura
A síndrome de Waardenburg tipo I (WS1) é uma doença pigmentar-auditiva que compreende perda auditiva neurossensorial congênita e distúrbios pigmentares da íris, cabelo e pele, juntamente com a distopia canthorum (deslocamento lateral da região interna da córnea). A perda auditiva em WS1, observada em aproximadamente 60% dos indivíduos afetados, é congênita, tipicamente não progressiva, seja unilateral ou bilateral, e neurossensorial. Mais comumente, a perda auditiva no WS1 é bilateral e profunda (> 100 dB). A maioria dos indivíduos com WS1 tem um topete branco ou um envelhecimento prematuro do cabelo do couro cabeludo antes dos 30 anos de idade. O topete branco clássico observado em aproximadamente 45% dos indivíduos é a anomalia de pigmentação capilar mais comum em WS1. Os indivíduos afetados podem ter heterocromia completa irídio, heterocromia parcial / segmentar ou irides azuis hipoplásicas ou brilhantes. O leucoderma congênito é freqüentemente visto na face, tronco ou membros.
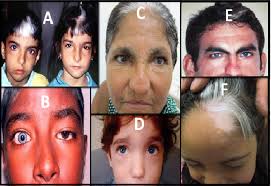


Diagnóstico:
O diagnóstico de WS1 é estabelecido na maioria dos indivíduos pelo exame físico por critérios clínicos, incluindo: perda auditiva neurossensorial, alterações pigmentares nos cabelos e olhos, distopia observada pelo cálculo do índice W e características faciais específicas. A identificação de uma variante patogênica PAX3 heterozigótica por meio de testes genéticos moleculares estabelece o diagnóstico se as características clínicas forem inconclusivas.
Tratamento das manifestações:
O manejo da perda auditiva depende de sua gravidade; implante coclear tem sido usado com sucesso em indivíduos com WS1.
Avaliação de parentes em risco:
Se a variante patogênica PAX3 específica da família é conhecida, o teste genético molecular de parentes em risco permite a triagem precoce daqueles com risco de perda auditiva.
Manejo da gravidez:
A suplementação de ácido fólico na gravidez é recomendada para mulheres com risco aumentado de ter uma criança com WS1 devido ao possível aumento do risco de defeitos do tubo neural em associação com a WS1.
Aconselhamento genético:
A síndrome de Waardenburg tipo I (WS1) é herdada de maneira autossômica dominante. A maioria dos probandos tem um pai afetado. Uma minoria de probandos não tem um pai afetado e presume-se que tenham WS1 como resultado de uma variante patogênica de novo. Prole de um indivíduo com WS1 tem 50% de chance de herdar a variante patogênica. Se a variante patogênica foi identificada em um membro da família afetado, o teste pré-natal para gravidez com risco aumentado pode estar disponível em um laboratório clínico que ofereça testes para essa doença / gene ou teste pré-natal personalizado. Embora este teste possa determinar se o feto herdou a variante patogênica PAX3, ele não pode determinar as manifestações clínicas ou sua gravidade.
Referência Bibliográfica:
Milunsky JM. Waardenburg Syndrome Type I. 2001 Jul 30 [Updated 2017 May 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1531/
Comentários