
Síndrome de Kabuki
- 14 de set. de 2018
- 2 min de leitura
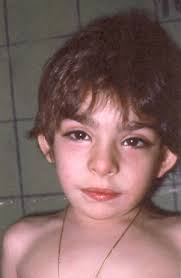
Também conhecida como Síndrome da Maquiagem de Kabuki, é caracterizada por características faciais típicas (fendas palpebrais alongadas com eversão do terço lateral da pálpebra inferior; sobrancelhas arqueadas e largas; columela curta com ponta nasal deprimida; orelhas grandes, proeminentes ou em concha), anomalias esqueléticas menores , persistência das almofadas dos dedos fetais, incapacidade intelectual leve a moderada e deficiência de crescimento pós-natal. Outros achados podem incluir: defeitos cardíacos congênitos, anomalias geniturinárias, lábio leporino e / ou palato, anomalias gastrintestinais incluindo atresia anal, ptose e estrabismo, e dentes muito espaçados e hipodontia. As diferenças funcionais podem incluir: aumento da suscetibilidade a infecções e distúrbios autoimunes, convulsões, anormalidades endocrinológicas, incluindo telarca prematura isolada em mulheres, problemas de alimentação e perda auditiva.


Diagnóstico:
O diagnóstico é primeiramente estabelecido pelos achados clínicos. O KS é causado por variantes patogênicas no KMT2D (anteriormente MLL2) ou KDM6A.
Tratamento das manifestações:
Aumento das mamadas e posicionamento após as refeições no tratamento do refluxo gastroesofágico; colocação de tubo de gastrostomia se as dificuldades de alimentação forem severas. Se as dificuldades cognitivas são evidentes, testes psicoeducacionais e serviços de educação especial para atender às necessidades individuais da criança. Avaliação por um pediatra ou psiquiatra do desenvolvimento se o comportamento sugerir transtornos do espectro do autismo. Tratamento antiepiléptico padrão para convulsões.
Prevenção de complicações secundárias:
O tratamento antibiótico profilático antes e durante qualquer procedimento (por exemplo, trabalho dentário) pode ser indicado para aqueles com defeitos cardíacos específicos.
Vigilância:
Monitore a altura, o peso e o perímetro cefálico a cada visita de criança e, no mínimo, anualmente. Os marcos do desenvolvimento devem ser seguidos com cada visita a crianças em bom estado. Monitore a visão e a audição anualmente.
Aconselhamento genético:
O KS relacionado ao KMT2D é herdado de maneira autossômica dominante. Cada criança de um indivíduo com KS relacionado ao KMT2D tem 50% de chance de herdar a variante patogênica. Até o momento, apenas seis indivíduos com variantes patogênicas ou deleções de KDM6A foram relatados; todos tiveram uma variante de novo comprovada ou aparente. Embora a herança ligada ao X seja teoricamente possível, não foram relatados casos familiares de SK resultantes de variantes patogênicas no gene KDM6A. A proporção de SK causada por variantes de novo é desconhecida, mas provavelmente é alta com base na experiência clínica. O diagnóstico pré-natal para gravidezes com risco aumentado é possível se a variante patogênica em um membro afetado da família for conhecida.
Referência bibliográfica:
Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. 2011 Sep 1 [Updated 2013 May 16]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK62111/
Comentários